
Computational Biophysics & Molecular Modeling
Samik Bose
Fixed-Term Assistant Professor
Department of Computational Mathematics, Science & Engineering
Michigan State University · East Lansing, Michigan, USA
Computational Biophysics & Molecular Modeling
Fixed-Term Assistant Professor
Department of Computational Mathematics, Science & Engineering
Michigan State University · East Lansing, Michigan, USA
About
I am a Fixed-Term Assistant Professor in the Department of Computational Mathematics, Science and Engineering at Michigan State University. Since joining the department in Fall 2024, I have taught several undergraduate and graduate courses, including Computational Medicine, Linear Algebra and Matrix Applications, Machine Learning in Molecular Dynamics, and Independent Research Study. I also continue my postdoctoral research in Computational Biophysics and Pharmacology under the mentorship of Prof. Alex Dickson (Biochemistry and Molecular Biology, MSU).
As an independent faculty member, my goal is to combine the complementary strengths of machine learning and theoretical physical chemistry to develop computational methods for pharmacologically relevant, long-timescale processes — augmenting biomedical and health research through molecular modeling. With my expertise in theoretical chemistry and my interest in drug discovery, I aim to provide a molecular basis for solving critical problems in human health and biology.
To that end, I work to strengthen the synergy between experiment and computation, drawing on facilities such as cryo-EM, NMR, and mass spectrometry. I currently collaborate with experimental scientists across medicinal chemistry (Prof. V. T. Karamyan, Oakland University), cryo-EM and structural biology (Dr. B. J. Orlando, MSU), pharmacology (Dr. K. S. S. Lee, MSU), and biochemistry and structural biology (Prof. A. A. Pioszak, University of Oklahoma Health Sciences Center). These collaborations, at the interface of chemistry, biology, and medicine, continually sharpen my view of the molecular-modeling tools the community needs.
Research
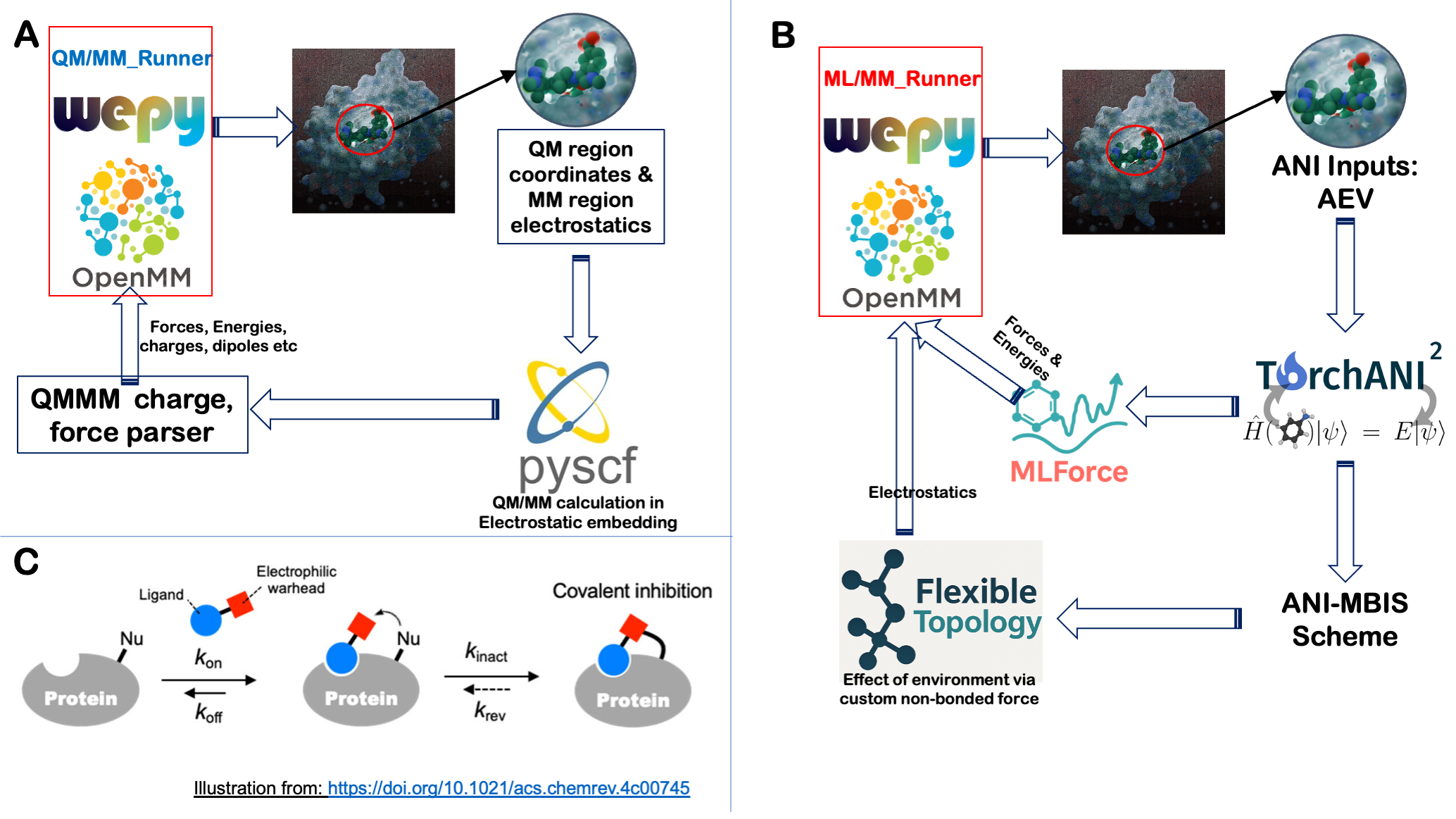
A weighted-ensemble framework (wepy + OpenMM) coupled to first-principles and machine-learning potentials: QM/MM in electrostatic embedding via PySCF, and ML/MM via TorchANI and MLForce with a Flexible Topology treatment of the environment.
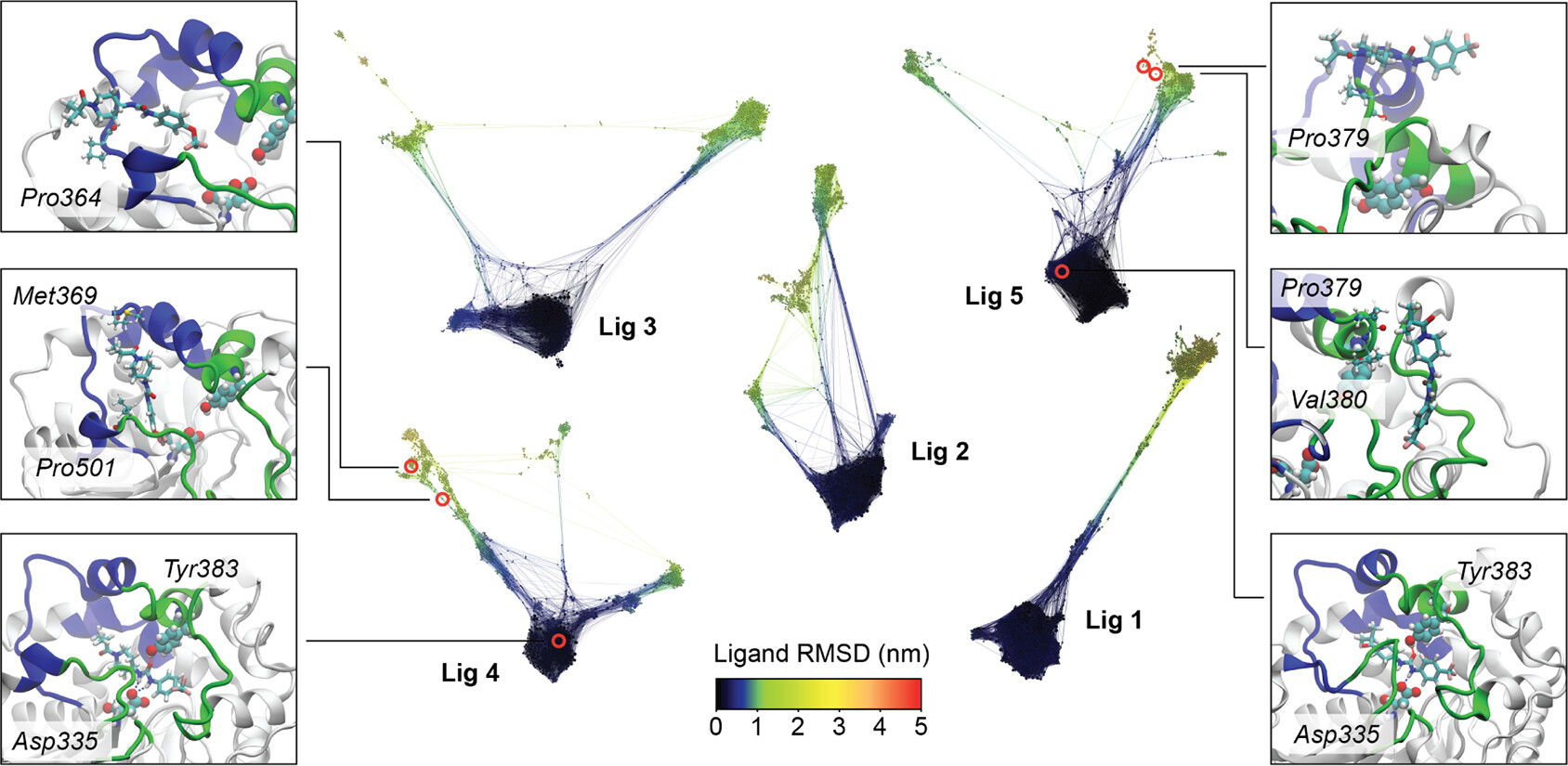
Inhibitor unbinding from soluble epoxide hydrolase (sEH): REVO-enhanced MD projected onto conformational-space networks.
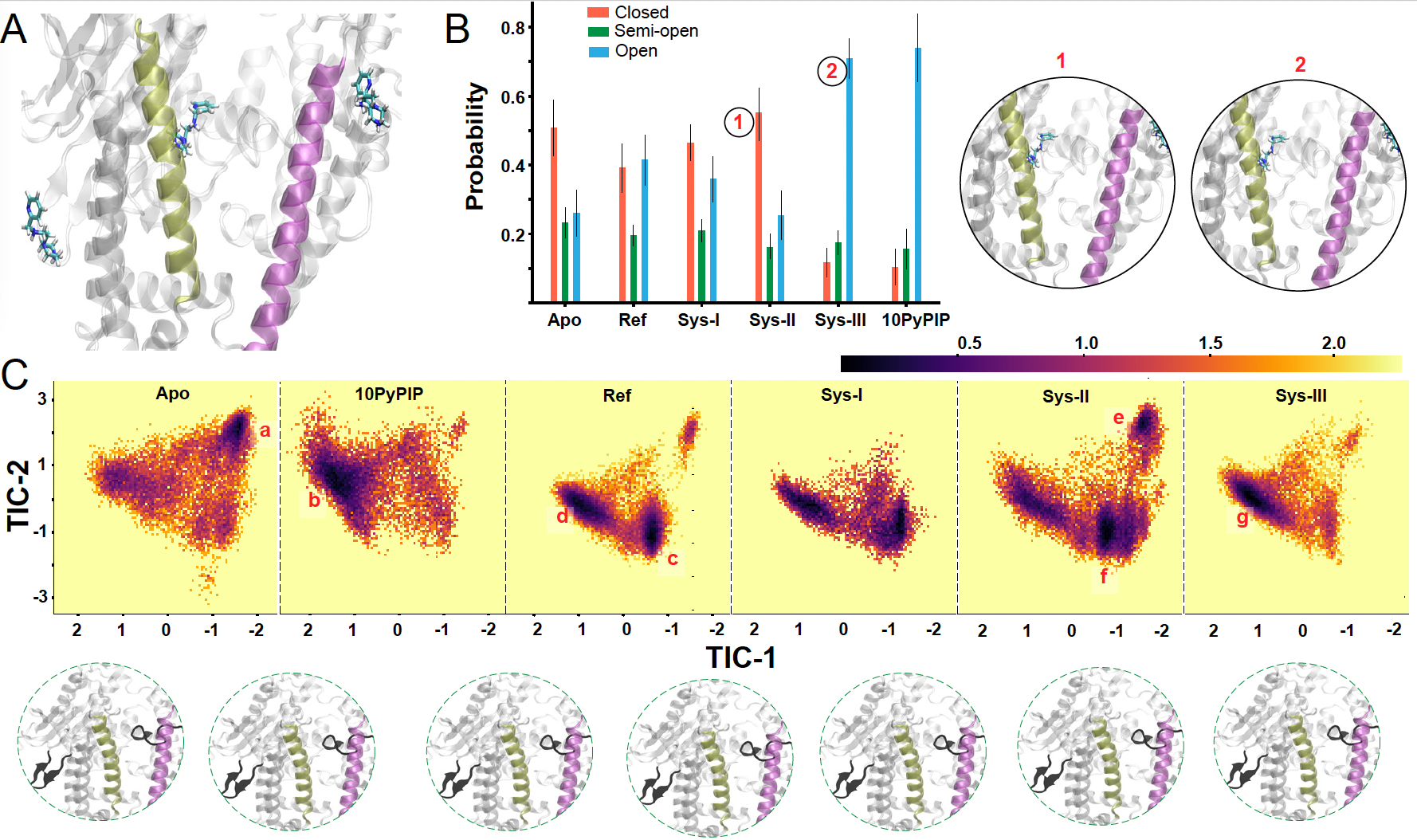
Effect of activator binding across different allosteric sites.
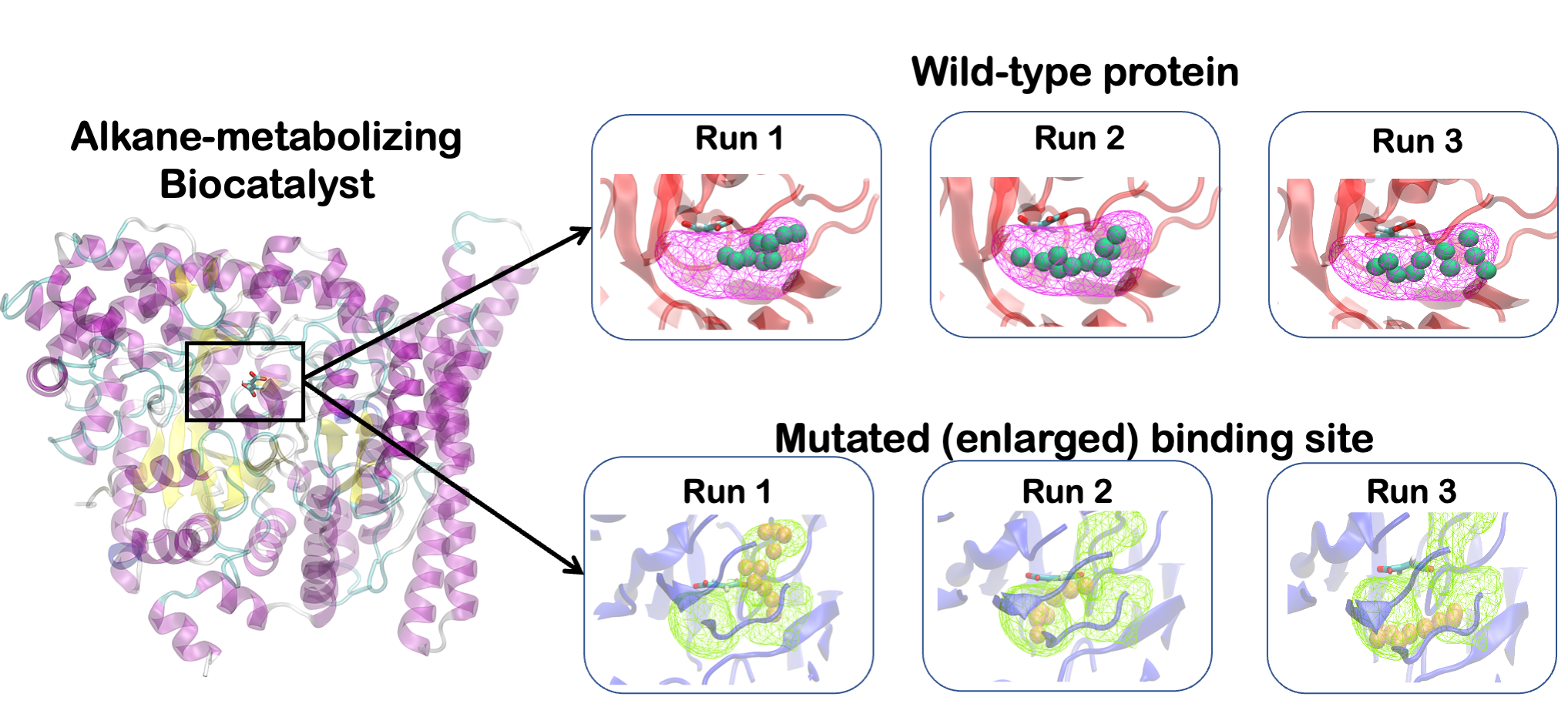
Keywords: computational chemistry · molecular dynamics · machine learning · enhanced sampling · weighted ensemble · Markov state modeling · QM/MM.
Publications
Teaching
Contact
The best way to reach me is by email at bosesami@msu.edu or samikbose20121990@gmail.com.